
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, affecting 1% of individuals over the age of 60.[1] Though the disease was first described more than 200 years ago, effective treatment options remain elusive. Clinical trials have revealed difficulties in translating results from animal models into human treatments. This highlights the necessity for basic researchers to carefully consider their animal models and experimental design.
In this article, we will discuss the pros and cons for the currently-available mouse models of PD. We will also review some of the most common behavioral tests for studying PD in rodent models.
Overview of Parkinson’s Disease
The most common symptoms of PD are movement problems, including rigidity, tremors, and bradykinesia (slowness of movement).[1] In addition, patients experience non-motor symptoms such as cognitive dysfunction and mood disorders.[2] PD is usually diagnosed after age 50 and is slightly more common in women than men. With the exception of a few rare varieties caused by a single genetic mutation, most cases of PD have no known cause.[1]
The pathological hallmark of PD is the presence of Lewy bodies in the brain. Lewy bodies are toxic fibrillar deposits made primarily from the α-synuclein protein, which accumulates inside of neurons and disrupts their proper function. Lewy bodies contribute to the loss of dopamine-producing neurons in a part of the brain called the substantia nigra, leading to the deterioration of motor control in PD. As the disease progresses, Lewy bodies spread to other regions of the brain and lead to the development of non-motor symptoms.[1]
Current therapies for PD cannot fully ameliorate the progression of the disease, and many, include considerable adverse side effects.[3] As a result, the development of effective PD treatments remains an active field of research in neurobiology.
Rodent Models of Parkinson’s Disease
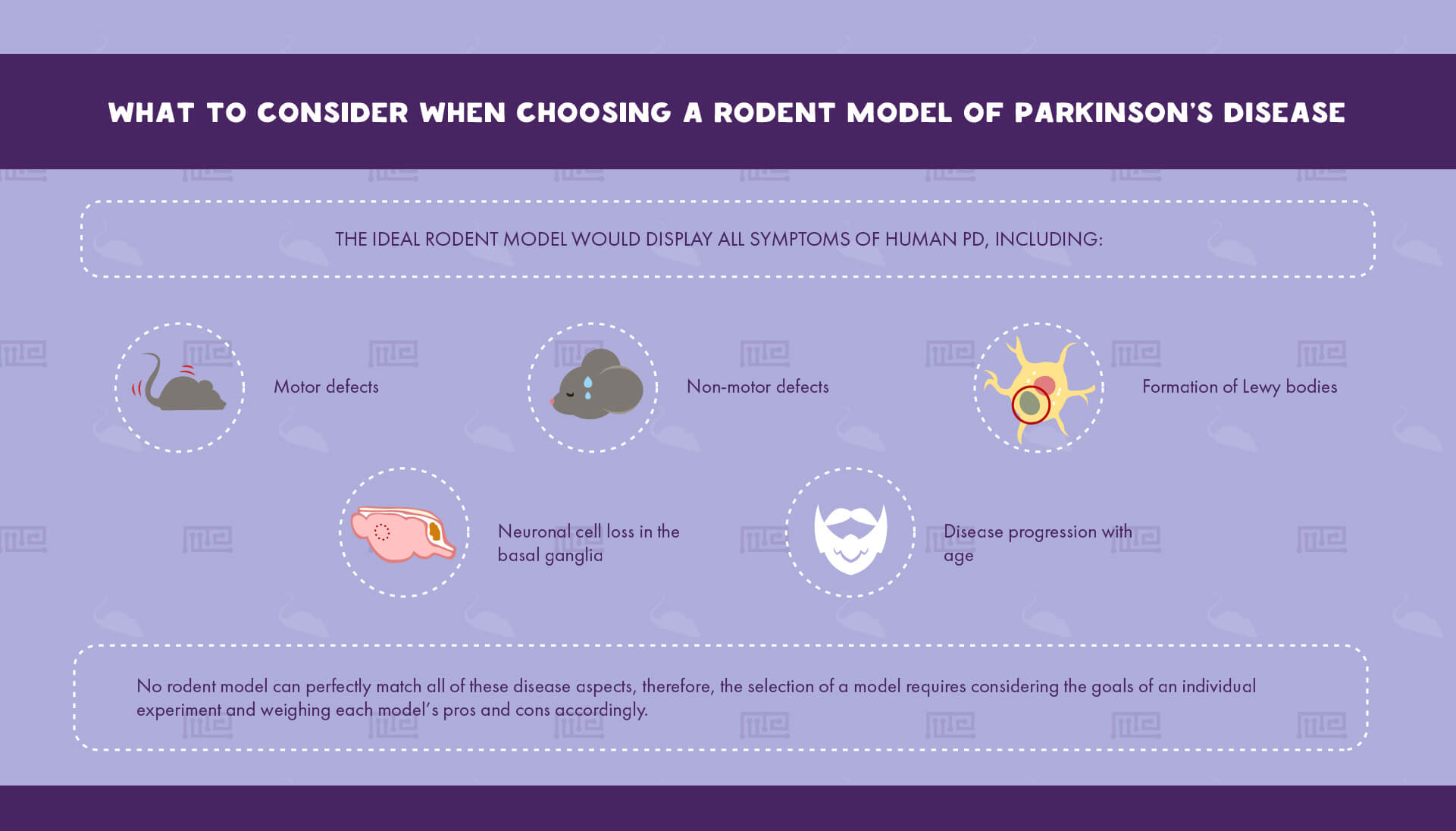
Determining which PD animal model to utilize is a critical step in planning an experiment that can dramatically affect its relevance to human biology. Rodents are the most common PD models due to their relatively low cost of maintenance and their ability to recapitulate many of PD’s key hallmarks.[4] The ideal rodent model would display all symptoms of human PD, including: [4]
- Motor defects
- Formation of Lewy bodies
- Neuronal cell loss in the basal ganglia
- Disease progression with age
- Non-motor defects
Since no rodent model can perfectly match all of these disease aspects, the selection of a model requires considering the goals of an individual experiment and weighing each model’s pros and cons accordingly.
In this section, we will provide an overview of different PD models to aid in the selection process. These models can be divided into three broad categories: [4, 5]
- Pharmacological models
- Herbicide and pesticide models
- Genetic models
Pharmacological Models
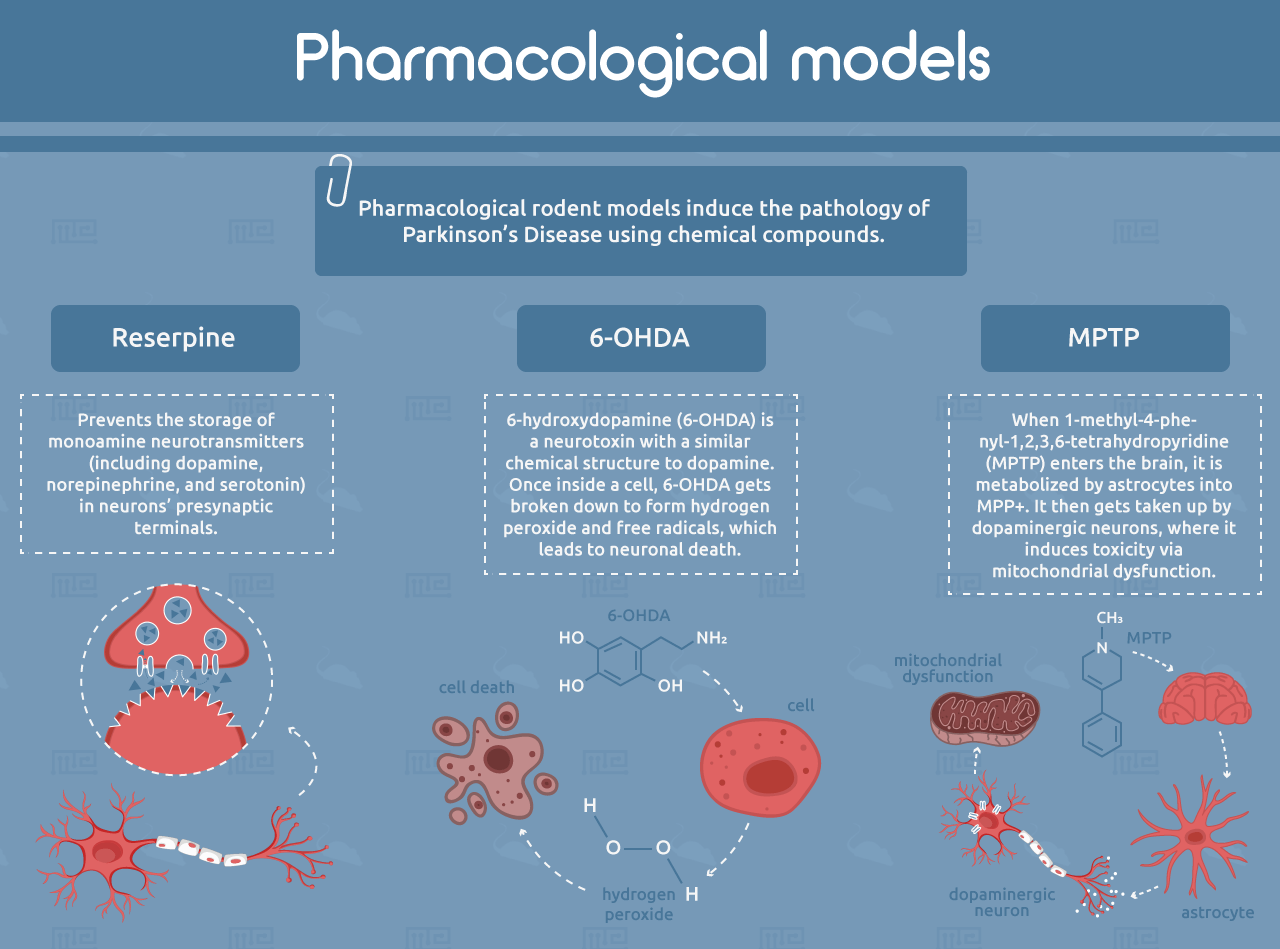
Reserpine
Pharmacological rodent models induce the pathology of PD using chemical compounds. One of the earliest examples is reserpine, which prevents the storage of monoamine neurotransmitters (including dopamine, norepinephrine, and serotonin) in neurons’ presynaptic terminals.[6, 7] This causes depletion of these neurotransmitters in the striatum, resulting in motor symptoms that resemble PD, including rigidity and slowness of movement. Reserpine rabbit models were used to discover the first dopaminergic PD drugs, including L-DOPA.[4, 5] However, the reserpine model has limited human relevance because of its broad depletion of monoamine neurotransmitters and the transient nature of its effects. It also does not show the formation of Lewy bodies or age-related progression.[4]
6-OHDA
6-hydroxydopamine (6-OHDA) is a neurotoxin with a similar chemical structure to dopamine. It can be taken up by dopamine transporters, which are expressed on the synapses of dopamine-producing neurons. 6-OHDA also has a high affinity for norepinephrine transporters, so it is often co-administered with a norepinephrine reuptake inhibitor in order to spare noradrenergic neurons.[8] Once inside the cell, 6-OHDA gets broken down to form hydrogen peroxide and free radicals, which leads to neuronal death.[9] The compound cannot cross the blood-brain barrier and must be administered by intracerebral injection. When administered to only one brain hemisphere, 6-OHDA allows each animal to serve as its own internal control by comparing treated and untreated hemispheres.[5] However, like with reserpine, the 6-OHDA model does not recapitulate Lewy body formation or progression with age.[4]
MPTP
Another common neurotoxic model of PD is 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). When MPTP enters the brain, it is metabolized by astrocytes into MPP+. It then gets taken up by dopaminergic neurons, where it induces toxicity via mitochondrial dysfunction.[10] Mice injected with MPTP recapitulate the motor defects of PD, but often to a milder extent than what is observed in primate models. Mice also develop very few α-synuclein-positive inclusions. It is important to note that mouse strains can vary widely in their sensitivity to MPTP, while rats are entirely resistant.[4, 5]
Pesticide and Herbicide Models
Paraquat and Maneb
Many pesticide and herbicide models for PD function similarly to the pharmacological models, in that they bear structural similarity to dopamine. One example is paraquat, a common agricultural herbicide. Mice treated with paraquat show α-synuclein accumulation in the substantia nigra.[11] However, since paraquat kills neurons via oxidative stress, its modeling of neuronal death in PD remains limited.[5] Paraquat is frequently administered in combination with a fungicide called maneb to enhance its deleterious effects.[12]
Rotenone
Rotenone is a combination of herbicide and pesticide that, like MPP+, preferentially targets dopaminergic neurons in the nigrostriatal pathway and induces accumulation of α-synuclein.[13] Its mechanism of neurotoxicity likely involves triggering the intracellular release of dopamine. [14] Because it is not dependent on the dopamine transporter, rotenone also exerts some toxicity on other neurotransmitter systems, although dopaminergic neurons are its primary targets.[4, 5] Additional complicating factors for the use of rotenone include high variability between individuals and the potential to induce systemic toxicity, leading to mortality.[15]
Trichloroethylene
A less well-characterized PD model is trichloroethylene (TCE), an industrial degreasing agent that is recognized as an environmental risk factor for PD.[16] Rats treated with TCE develop α-synuclein accumulation, loss of nigrostriatal dopaminergic neurons, and other PD hallmarks. However, they do not show significant depletion of dopamine.[17, 18] Additional research is needed to understand the translational relevance of the TCE model.[4]

Genetic Models
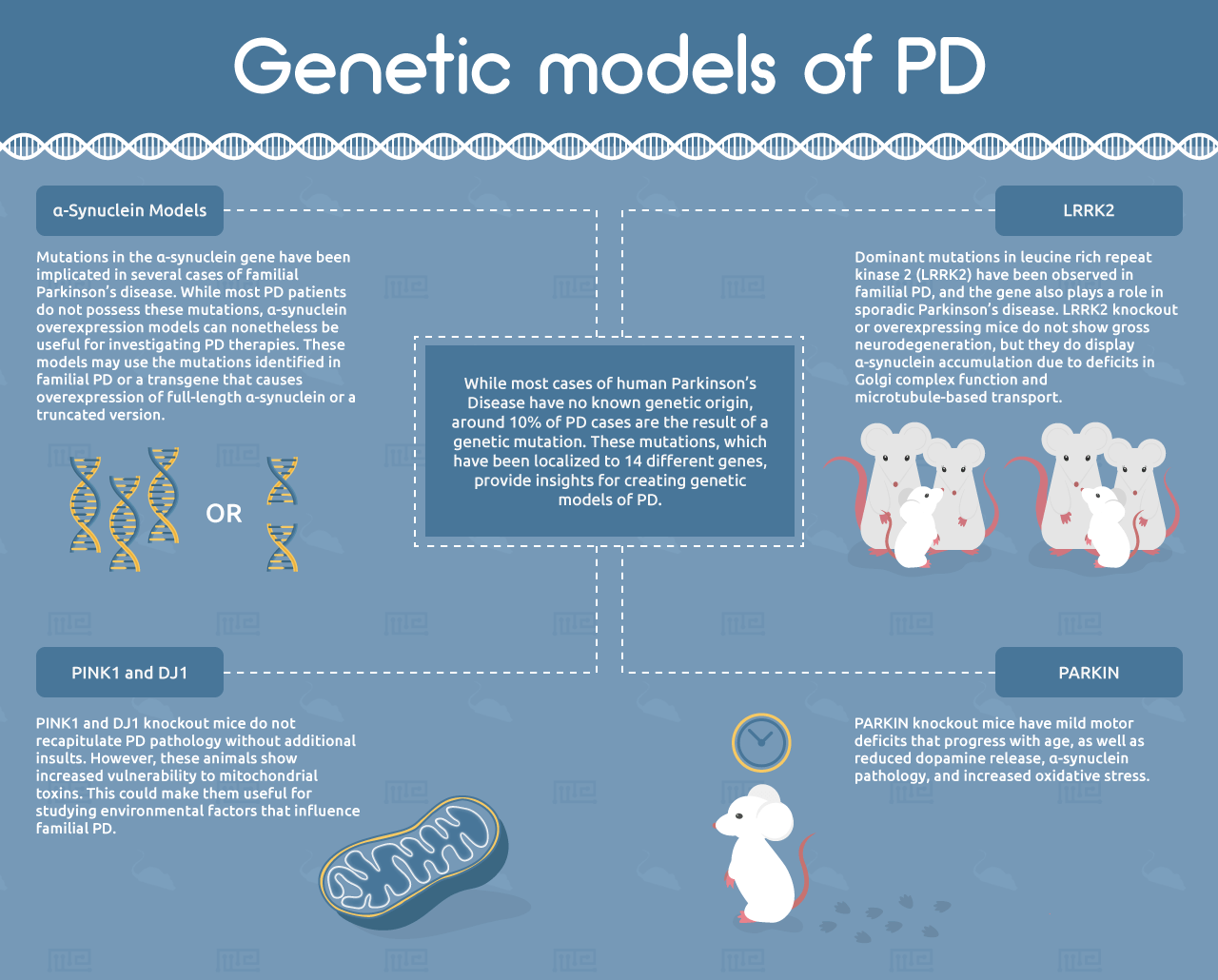
While most cases of human PD have no known genetic origin, around 10% of PD cases are the result of a genetic mutation.[19] These mutations, which have been localized to 14 different genes, provide insights for creating genetic models of PD.[3]
α-Synuclein Models
Mutations in the α-synuclein gene have been implicated in several cases of familial Parkinson’s disease. While most PD patients do not possess these mutations, α-synuclein overexpression models can nonetheless be useful for investigating PD therapies. These models may use the mutations identified in familial PD or a transgene that causes overexpression of full-length α-synuclein or a truncated version.[5] An important advantage of these models over the pharmacological and toxicological systems is that they exhibit progressive accumulation of α-synuclein with age.[20] However, most of the α-synuclein deposits in these mice do not have the characteristic fibrillar structure of Lewy bodies.[21] Another important limitation of α-synuclein-overexpressing mice is that they do not exhibit neuronal cell loss in the substantia nigra, suggesting that α-synuclein is not the sole driver of PD.[4, 5]
α-synuclein transgenes can be targeted via a selective promoter that determines in which cell types they are expressed. A commonly-used promoter is the tyrosine hydroxylase promoter, which targets dopaminergic neurons.[5] The pathology of this model is restricted to the nigrostriatal system, and the mice do not show the widespread α-synuclein accumulation that is seen in human PD. An alternative option is the platelet-derived growth factor-β (PDGF-β) promoter, which creates a broader distribution of α-synuclein overexpression. However, neuronal death and motor symptoms have not been observed in this model.[5, 22]
Prion protein promoter models overexpress α-synuclein in the nigrostriatal system and show severe motor dysfunction, but the transgene is also highly expressed in neurons not typically affected by PD, including spinal cord and brainstem motor neurons.[23] In contrast, the Thy-1 promoter widespread α-synuclein accumulation without affecting motor neurons. Notably, Thy-1 models have a late onset, or in some cases an absence, of motor deficits.[5] Another option is the calcium/calmodulin-depending protein kinase IIα (CaM) promoter, which allows for the temporal control of gene expression using the tetracycline transactivator system.[24] The CaM promoter/α-synuclein model can be useful for studying the early stages of PD pathology.[5]
PARKIN
Numerous other genetic models of PD have been generated based on mutations observed in familial PD. These include mutations in PARKIN, a ubiquitin E3 ligase involved in mitochondrial homeostasis. PARKIN knockout mice have mild motor deficits that progress with age, as well as reduced dopamine release, α-synuclein pathology, and increased oxidative stress. However, they do not show loss of dopaminergic neurons in the nigrostriatal system.[5] Notably, the Q311X PARKIN mutation has been shown to exhibit progressive nigrostriatal cell death and other PD hallmarks, suggesting this could be a very useful PD model.[25]
PINK1 and DJ1
Similarly to PARKIN, recessive mutations in the PINK1 and DJ1 genes have been observed in familial PD. PINK1 and DJ1 knockout mice do not recapitulate PD pathology without additional insults. However, these animals show increased vulnerability to mitochondrial toxins. This could make them useful for studying environmental factors that influence familial PD.[5]
LRRK2
Dominant mutations in leucine rich repeat kinase 2 (LRRK2) have been observed in familial PD, and the gene also plays a role in sporadic Parkinson’s disease.[5] LRRK2 knockout or overexpressing mice do not show gross neurodegeneration, but they do display α-synuclein accumulation due to deficits in Golgi complex function and microtubule-based transport.[26] These models also show enhanced pathological progression induced by α-synuclein overexpression. Mice overexpressing the G2019S-mutant version of LRRK2 show an age-dependent decrease in striatal dopamine and reduced adult neurogenesis.[27-29] The translational relevance of the LRRK2 model has not yet been investigated, but the involvement of LRRK2 in both familial and sporadic PD make this model an attractive candidate.[5]
Viral-Mediated Gene Delivery
An alternative to generating knockout or transgenic mice is to utilize viral-mediated gene delivery. This system utilizes viruses to insert novel DNA sequences into the genome of a host cell. Previous studies have used viral vectors to overexpress wild-type or mutant α-synuclein in the substantia nigra. The animals develop fibrillar deposits resembling Lewy bodies that form gradually over a period of weeks.[30] This method has also been utilized for PARKIN overexpression, which showed encouraging results for neuroprotection in PD.[31, 32]
Viral vectors allow for a high degree of temporal and locational specificity, enabling experimenters to induce transgene expression in adult wild-type animals. They also allow animals to be unilaterally transduced and serve as their own control, which is not possible for knockout or transgenic animals.[30] However, viral vectors come with drawbacks. The procedure is technically challenging, requiring a direct injection into the brain. The level of transgene expression may vary considerably between animals, and even between different neurons in the same animal.[30] One cohort reported that only 25-30% of mice treated with an α-synuclein-encoding vector developed noticeable deficits.[33] These variable results should be weighed against the advantages of temporally-controlled gene delivery.
Clinical Relevance of Parkinson’s Disease Models
The ability of an animal model to inform human clinical studies is of key importance when designing PD experiments. Among the PD rodent models, their most common critique is the lack of Lewy body formation. Studies of primate models have helped to explain this observation. In monkeys treated with MPTP, α-aggregation mainly occurs in neurons expressing neuromelanin, a pigment that is found in dopaminergic neurons of primates but not rodents.[34] This finding suggests that Lewy bodies may result from a unique mechanism in primate dopaminergic neurons. The inability to replicate a key pathological feature of PD is a caution against the clinical relevance of these neurotoxic models.[30]
In addition to the limitations of rodent models, poor experimental design can further widen the gap between animal research and human trials. For instance, approximately half of all PD drug candidates tested in rodents are administered simultaneously or prior to neurotoxin administration.[30] This is in stark contrast to human PD, where treatment is not initiated until after months or years of progressive neurodegeneration. Incorporating clinically-driven experimental design will help to enhance the relevance of these studies for human trials.[30]
Behavioral Assessments for Parkinson’s Disease Rodent Models
In addition to considering the pros and cons of different PD rodent models, selecting the appropriate assay is an equally important factor for designing effective experiments. Behavioral assessments are a useful tool to evaluate the motor and non-motor symptoms of PD in animal models. PD behavioral assessments can be divided into three broad categories: [35]
- Motor evaluation
- Neuropsychological evaluation
- Hyposmia evaluation
Motor Evaluation
Numerous motor tests are available to characterize the broad spectrum of PD motor deficits. For example, tests that quantify the animal’s free movements (either in the home cage or an open field) are frequently used to glean a general assessment of motor function. The cylinder test can also be used to assess spontaneous movement. In this test, the animal is placed inside a cylinder for ten minutes while its number of forepaw touches to the cylinder is counted.[35]
Other kinds of tests are designed to characterize specific types of motor dysfunction. The pole test is commonly used to evaluate bradykinesia (slowness of movement). In this test, the animal is placed facing upward on a vertical pole, and the time to reverse itself and climb down is measured.[36] The beam test, in which the animal walks along a narrow balance beam, evaluates motor coordination and gait. A more complex version of the beam test called the Michigan Complex Motor Control Test substitutes a rotating rod for the balance beam to measure posture and precise motor control.[37, 38]
An additional class of motor tests relies on observing walking behavior. Active walking tests, including the balance beam test, gait test, and the grid test, allow the animal to walk freely, while passive walking tests force the animal to move by placing them on a treadmill, rotarod, or automated running wheel.[35, 39, 40] For evaluating finer motor activity such as forelimb grasping, the stairway test or the vermicelli handling test may be used.[37, 41, 42]
Neuropsychological Evaluation
Neuropsychological tests analyze the non-motor symptoms of PD, including depression, anxiety, and cognitive impairment. PD models present unique challenges in monitoring neuropsychological symptoms, and particular care must be taken to avoid the introduction of bias due to motor deficits.
While depression may seem difficult to evaluate in nonhuman animals, experiments such as the sucrose-consumption test allow for easy measurement of depressive-like symptoms. An animal that, when allowed unlimited consumption of a sucrose solution for a period of several days, shows reduced intake of the solution is said to exhibit anhedonia (a reduced ability to feel pleasure).[43] Other common measures of depression-like symptoms, including the forced swim test [44] and the tail suspension test,[45] are not advisable for PD models because their limited movements are a result of PD rather than depression.[35]
Anxiety is a common symptom of human PD. One method of measuring anxiety in rodent models is the open field test, where the animal is allowed to move freely within a room.[46] Video camera monitoring allows for the animal’s motion to be quantified. A similar experiment is the elevated-plus maze test, where the animal explores a plus-shaped maze. Since rodents prefer to avoid open spaces, the proportion of time that an animal spends in the open arms of the maze can be used to evaluate anxiety.[47, 48]
The final type of neuropsychological test used in PD models is for cognitive impairment. The motor defects seen in PD rodent models can make cognitive impairment testing more complicated. For example, the Morris water maze test, where a mouse has to remember the location of a hidden platform, can be impacted by bradykinesia in PD animals.[35, 49] Other types of mazes with less reliance on motor function may be more effective for evaluating spatial memory in PD models. Alternative to the Morris water maze includes the radial arm maze, the Y-maze, the T-maze, and the Barnes maze.[35, 50-52] In addition, the novel object recognition testis widely used in PD models and is sensitive enough to detect mild impairments.[35, 53]
Hyposmia
Hyposmia, the reduced ability to smell, is a common symptom of early-stage PD that is also seen in many PD rodent models.[54] Tests for anosmia frequently evaluate the animal’s ability to locate hidden food using olfaction.[55, 56] However, the motor defects seen in PD models may confound these experiments.[35] A preferable alternative involves the placement of two odorants in a radial maze, where one odorant is associated with water as a reward. The animal’s preference for the reward-associated smell serves as a measure of olfactory ability. This test was previously used to evaluate hyposmia in the 6-OHDA model.[57]
Conclusion
No animal model can perfectly recapitulate all the symptoms and pathologies found in human PD. Therefore, it is necessary for the experimenter to carefully consider the questions they wish to answer and weigh the benefits and drawbacks of each model accordingly. After selecting the appropriate PD model, an equally important step is to determine which experimental test(s) to employ for evaluating the animals’ motor, neuropsychological, or olfactory defects.
For our apparatuses used in assessing motor function for conditions such as Parkinson’s disease, see our activity range here.
References
- Tysnes, O.B. and A. Storstein, Epidemiology of Parkinson’s disease.J Neural Transm (Vienna), 2017. 124(8): p. 901-905.
- Rana, A.Q., et al., Parkinson’s disease: a review of non-motor symptoms.Expert Rev Neurother, 2015. 15(5): p. 549-62.
- Schapira, A.H. and E. Tolosa, Molecular and clinical prodrome of Parkinson disease: implications for treatment.Nat Rev Neurol, 2010. 6(6): p. 309-17.
- Antony, P.M., N.J. Diederich, and R. Balling, Parkinson’s disease mouse models in translational research.Mamm Genome, 2011. 22(7-8): p. 401-19.
- Blesa, J., et al., Classic and new animal models of Parkinson’s disease.J Biomed Biotechnol, 2012. 2012: p. 845618.
- Carlsson, A., M. Lindqvist, and T. Magnusson, 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists.Nature, 1957. 180(4596): p. 1200.
- Steg, G., Alpha-rigidity in reserpinized rats.Experientia, 1964. 20(2): p. 79-80.
- Perese, D.A., et al., A 6-hydroxydopamine-induced selective parkinsonian rat model.Brain Res, 1989. 494(2): p. 285-93.
- Ungerstedt, U., T. Ljungberg, and G. Steg, Behavioral, physiological, and neurochemical changes after 6-hydroxydopamine-induced degeneration of the nigro-striatal dopamine neurons.Adv Neurol, 1974. 5: p. 421-6.
- Langston, J.W., et al., Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis.Science, 1983. 219(4587): p. 979-980.
- Manning-Bog, A.B., et al., The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein.J Biol Chem, 2002. 277(3): p. 1641-4.
- Thiruchelvam, M., et al., The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson’s disease.J Neurosci, 2000.20(24): p. 9207-14.
- Cannon, J.R., et al., A highly reproducible rotenone model of Parkinson’s disease.Neurobiol Dis, 2009. 34(2): p. 279-90.
- Inden, M., et al., Parkinsonian rotenone mouse model: reevaluation of long-term administration of rotenone in C57BL/6 mice.Biol Pharm Bull, 2011. 34(1): p. 92-6.
- Ravenstijn, P.G., et al., The exploration of rotenone as a toxin for inducing Parkinson’s disease in rats, for application in BBB transport and PK-PD experiments.J Pharmacol Toxicol Methods, 2008. 57(2): p. 114-30.
- Goldman, S.M., Trichloroethylene and Parkinson’s disease: dissolving the puzzle.Expert Rev Neurother, 2010. 10(6): p. 835-7.
- Liu, P., et al., G protein-coupled receptor kinase 5, overexpressed in the alpha-synuclein up-regulation model of Parkinson’s disease, regulates bcl-2 expression.Brain Res, 2010. 1307: p. 134-41.
- Liu, M., et al., Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats.J Neurochem, 2010. 112(3): p. 773-83.
- Dauer, W. and S. Przedborski, Parkinson’s disease: mechanisms and models.Neuron, 2003. 39(6): p. 889-909.
- Fernagut, P.O. and M.F. Chesselet, Alpha-synuclein and transgenic mouse models.Neurobiol Dis, 2004. 17(2): p. 123-30.
- Maries, E., et al., The role of alpha-synuclein in Parkinson’s disease: insights from animal models.Nat Rev Neurosci, 2003. 4(9): p. 727-38.
- Yacoubian, T.A., et al., Transcriptional dysregulation in a transgenic model of Parkinson disease.Neurobiol Dis, 2008. 29(3): p. 515-28.
- Magen, I. and M.F. Chesselet, Genetic mouse models of Parkinson’s disease The state of the art.Prog Brain Res, 2010. 184: p. 53-87.
- Lim, Y., et al., Forebrain overexpression of alpha-synuclein leads to early postnatal hippocampal neuron loss and synaptic disruption.Exp Neurol, 2010. 221(1): p. 86-97.
- Lu, X.H., et al., Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein.J Neurosci, 2009. 29(7): p. 1962-76.
- Lin, X., et al., Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein.Neuron, 2009. 64(6): p. 807-27.
- Li, X., et al., Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S.J Neurosci, 2010. 30(5): p. 1788-97.
- Melrose, H.L., et al., Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice.Neurobiol Dis, 2010. 40(3): p. 503-17.
- Winner, B., et al., Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice.Neurobiol Dis, 2011. 41(3): p. 706-16.
- Bezard, E., et al., Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies.Mov Disord, 2013.28(1): p. 61-70.
- Vercammen, L., et al., Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson’s disease.Mol Ther, 2006. 14(5): p. 716-23.
- Lo Bianco, C., et al., Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease.Proc Natl Acad Sci U S A, 2004. 101(50): p. 17510-5.
- Kirik, D., et al., Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system.J Neurosci, 2002. 22(7): p. 2780-91.
- Purisai, M.G., et al., Alpha-synuclein expression in the substantia nigra of MPTP-lesioned non-human primates.Neurobiol Dis, 2005. 20(3): p. 898-906.
- Asakawa, T., et al., Animal behavioral assessments in current research of Parkinson’s disease.Neurosci Biobehav Rev, 2016. 65: p. 63-94.
- Ogawa, N., et al., A simple quantitative bradykinesia test in MPTP-treated mice.Res Commun Chem Pathol Pharmacol, 1985. 50(3): p. 435-41.
- Kucinski, A., et al., Modeling fall propensity in Parkinson’s disease: deficits in the attentional control of complex movements in rats with cortical-cholinergic and striatal-dopaminergic deafferentation.J Neurosci, 2013. 33(42): p. 16522-39.
- Kucinski, A., et al., Modeling falls in Parkinson’s disease: Slow gait, freezing episodes and falls in rats with extensive striatal dopamine loss.Behav Brain Res, 2015. 282: p. 155-64.
- Holmes, M.M., et al., Adult hippocampal neurogenesis and voluntary running activity: circadian and dose-dependent effects.J Neurosci Res, 2004. 76(2): p. 216-22.
- Rozas, G., M.J. Guerra, and J.L. Labandeira-Garcia, An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism.Brain Res Brain Res Protoc, 1997. 2(1): p. 75-84.
- Simiand, J., P.E. Keane, and M. Morre, The staircase test in mice: a simple and efficient procedure for primary screening of anxiolytic agents.Psychopharmacology (Berl), 1984. 84(1): p. 48-53.
- Montoya, C.P., et al., The “staircase test”: a measure of independent forelimb reaching and grasping abilities in rats.J Neurosci Methods, 1991. 36(2-3): p. 219-28.
- Papp, M., P. Willner, and R. Muscat, An animal model of anhedonia: attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress.Psychopharmacology (Berl), 1991. 104(2): p. 255-9.
- Porsolt, R.D., M. Le Pichon, and M. Jalfre, Depression: a new animal model sensitive to antidepressant treatments.Nature, 1977. 266(5604): p. 730-2.
- Steru, L., et al., The tail suspension test: a new method for screening antidepressants in mice.Psychopharmacology (Berl), 1985. 85(3): p. 367-70.
- Hall, C.S., Emotional behavior in the rat. I. Defecation and urination as measures of individual differences in emotionality.Journal of Comparative Psychology, 1934. 18(3): p. 385-403.
- Graeff, F.G., et al., Role of the amygdala and periaqueductal gray in anxiety and panic.Behav Brain Res, 1993. 58(1-2): p. 123-31.
- Holmes, A., et al., Behavioral profile of wild mice in the elevated plus-maze test for anxiety.Physiol Behav, 2000. 71(5): p. 509-16.
- Morris, R., Developments of a water-maze procedure for studying spatial learning in the rat.Journal of neuroscience methods, 1984. 11(1): p. 47-60.
- Hritcu, L. and A. Ciobica, Intranigral lipopolysaccharide administration induced behavioral deficits and oxidative stress damage in laboratory rats: relevance for Parkinson’s disease.Behav Brain Res, 2013. 253: p. 25-31.
- Hall, K., et al., Behavioural deficits in transgenic mice expressing human truncated (1-120 amino acid) alpha-synuclein.Exp Neurol, 2015. 264: p. 8-13.
- Ramos-Moreno, T., C.G. Castillo, and A. Martinez-Serrano, Long term behavioral effects of functional dopaminergic neurons generated from human neural stem cells in the rat 6-OH-DA Parkinson’s disease model. Effects of the forced expression of BCL-X(L).Behav Brain Res, 2012. 232(1): p. 225-32.
- Sik, A., et al., Performance of different mouse strains in an object recognition task.Behav Brain Res, 2003. 147(1-2): p. 49-54.
- Xiao, Q., S. Chen, and W. Le, Hyposmia: a possible biomarker of Parkinson’s disease.Neurosci Bull, 2014. 30(1): p. 134-40.
- Kurtenbach, S., et al., Olfaction in three genetic and two MPTP-induced Parkinson’s disease mouse models.PLoS One, 2013. 8(10): p. e77509.
- Yang, M. and J.N. Crawley, Simple behavioral assessment of mouse olfaction.Curr Protoc Neurosci, 2009. Chapter 8: p. Unit 8 24.
- Valle-Leija, P. and R. Drucker-Colin, Unilateral olfactory deficit in a hemiparkinson’s disease mouse model.Neuroreport, 2014. 25(12): p. 948-53.