
CRISPR, Genes, and Behavior
Through CRISPR gene-editing, researchers can carefully study the interaction between genes and behavior in a way that is more precise than what has been used before. Using this tool, researchers can add, remove or even alter genetic material in specific, targeted locations. Thus, researchers can manipulate genes in healthy or even disease model mice/rats and study the subsequent effects on behavior.
In this article, we will cover a few experiments that have used CRISPR gene-editing in mice or rats. But, first, let’s provide a bit of a background about what CRISPR gene-editing is and how it works.
What is CRISPR?
As mentioned previously, CRISPR is a targeted genome-editing tool that offers researchers control and precision.
Oftentimes, this technique is also referred to as CRISPR/Cas9 gene-editing. Cas9 is included in the phrase because it is a key protein that makes this technique possible.
CRISPR stands for ‘Clustered Regularly Interspaced Short Palindromic Repeats’. Together with Cas proteins, they are a part of the CRISPR/Cas9 immune system that is found naturally occurring in prokaryotic organisms like archaea and bacteria.[1] This DNA-encoded and RNA-mediated immune system is able to recognize, target, and degrade any exogenous nucleic acid. Eventually, researchers were able to modify it in order to create a handy gene-editing system for research purposes.
CRISPR/Cas9: The gene-editing system
This gene-editing system has several crucial components, including:
- Cas9: the CRISPR associated protein 9, more simply known as ‘Cas9,’ is a DNA endonuclease enzyme which has DNA site recognition and can also cleave (thus, modify) DNA where necessary. A few variants of Cas9 include single-strand nicking, DNA binding, and double-strand breaking.
- tracrRNA: will bind to crRNA and form an active complex.
- crRNA: is a region that binds with tracrRNA and locates the section of interest in the host DNA, thus forming an active complex.
- sgRNA: sgRNA, or ‘single-guide RNA,’ is the combined RNA that is made up of tracrRNA and at least one crRNA.
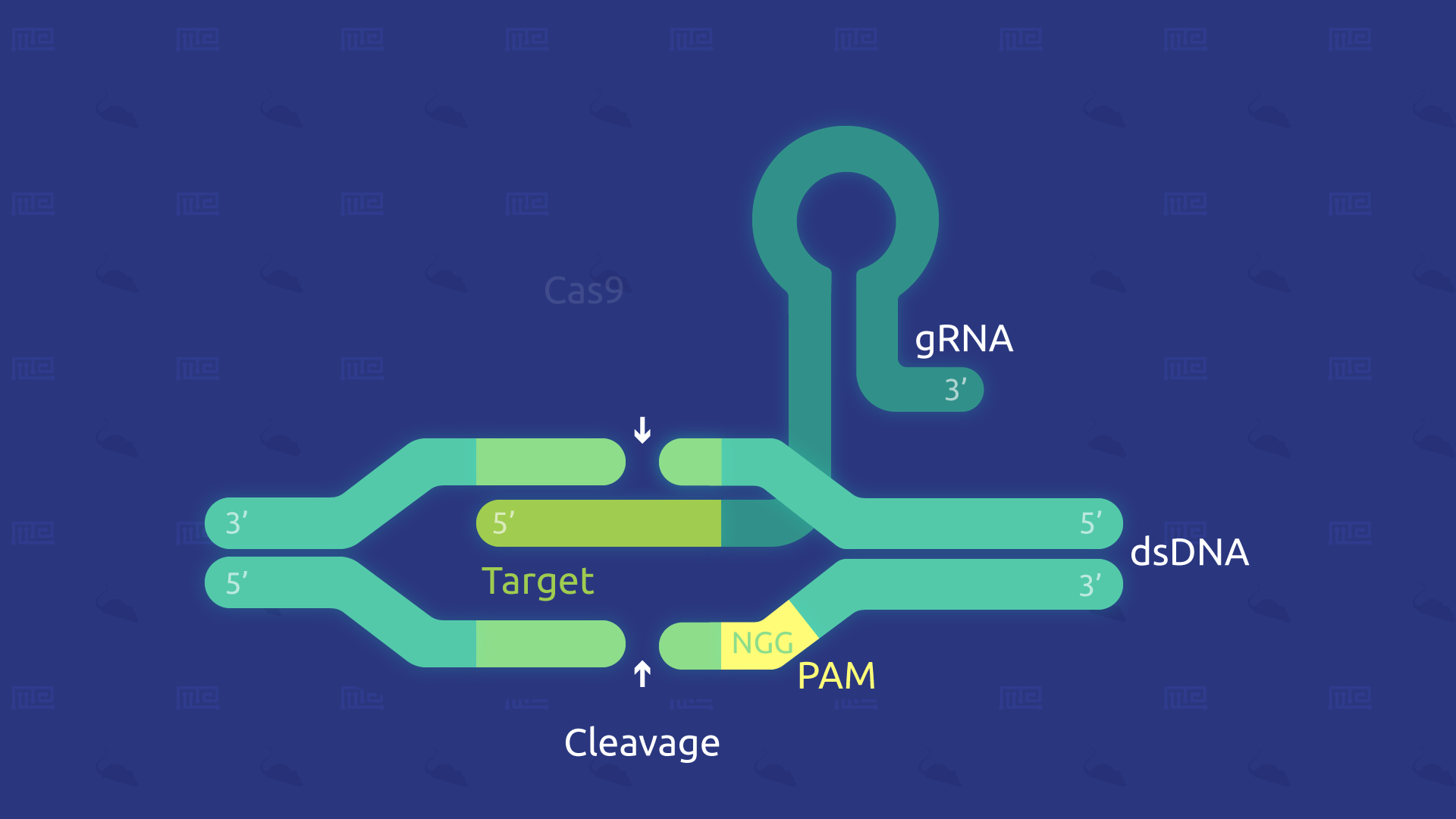
Together, these components are largely what drive the mechanisms involved in creating the desired animal models. Of course, there are other possible variations to this advanced genome-editing technique.
Shortcomings of CRISPR
Even though CRISPR can be targeted, precise, and applied to almost any behavioral research problem, there are still some pitfalls that are associated with this technique. One of the major issues is off-target effects like mutagenesis. So, even though CRISPR/Cas9 genome editing is a targeted, specific technique, there can be side-effects in the genome with regards to what happens beyond the targeted location. This is problematic because it doesn’t allow for scientists to have control over every aspect of the study, often leading to unpredictable outcomes.
Rodent Studies Using CRISPR
We will cover the following mini-case studies on how CRISPR/Cas9 is utilized in behavioral research and highlight the subsequent behavioral outcomes:
- Neurofibromatosis Induction in Rats with CRISPR/Cas9
- Ameliorating Huntington’s Disease Neurotoxicity with CRISPR/Cas9
- Using CRISPR/Cas9 to Understand Astrocyte Function
- Modeling Fragile X Syndrome in Rats with CRISPR
- Using CRISPR in Mice to Induce Rett Syndrome and Bioluminescence Imaging
Neurofibromatosis Induction in Rats with CRISPR/Cas9
Neurofibromatosis type 1 (NF1) is a rare autosomal dominant disease that has been linked to NF1 gene mutations. This condition is described by pigmentation (skin coloring), growth of benign or cancerous tumors throughout the body, and pain. People with NF1 face many other complications, including cognitive problems, hypertension, macrocephaly (an abnormally large head), scoliosis (abnormal spine curvature), and higher risk of developing leukemia. Worldwide studies have reported that one of the major complains NF1 patients have is pain, compromising their quality of life. However, the mechanisms behind the chronic pain they feel remain unclear.[2]
Studies using NF1+/- mice have identified the potential reason why NF1 patients experience such severe pain. Studies using NF1 haploinsufficient mice have revealed that sensitization of ion channels may be involved in the severe pain that NF1 patients suffer. But, the issue with using such a mouse model is that behavioral assessments lead to mixed findings regarding the function of NF1 to feeling pain. Mixed findings are due to the variability that mice exhibit with regards to stable pain stimuli. Some experimental mice demonstrate higher sensitivity to painful stimuli while other mice of the same strain show lower sensitivity to the same intensity of pain stimuli.
The CRISPR/Cas9 gene-editing system is a solution to this problem since it can be used to produce a rat model with NF1-related pain.[2] This model can be accomplished via spinal drug delivery of Nf1 sgRNAs. Such an induction method is less likely to produce damage to the surrounding spinal nerves in rats since rats are bigger than mice. Thus, injecting sgRNAs daily for the consecutive span of 3 days will lead to the induction of neurofibromatosis via CRISPR gene-editing. In the next section, we will see how behavior is assessed in the neurofibromatosis CRISPR/Cas9 model.
Assessing Behavior in Neurofibromatosis NF1 Rats
Using a Hargreaves Test Apparatus, the pain responses of NF1 rats can be quantified. Via controlled exposure to heat emitted from this apparatus, the time it takes for the rat to withdraw its paw is measured in seconds.
Both males and female NF1 rats will show a decreased withdrawal latency in the Hargreaves test when compared to control mice. Male NF1 rats may be more sensitive overall since they begin showing a dramatic decrease in withdrawal latency much sooner than females.
NF1 males will begin to show a statistically significant reduction of withdrawal latency just 3 days after the induction process has been completed. Females start to show meaningful differences after 6 days have passed after induction has been completed. Furthermore, while control rats have a withdrawal latency of about 20 seconds, NF1 rats have a withdrawal latency of about 10 seconds.
Interestingly, CRISPR-induced neurofibromatosis does not increase levels of anxiety in the experimental rats. In fact, when subjected to the Elevated Plus Maze, NF1 rats will not show anxiety levels that are statistically different from control mice. Such a finding is experimentally desirable because it means that the CRISPR/Cas9 approach is useful in creating a reductionist model that focuses exclusively on the relationship between pain and neurofibromatosis. Thus, anxiety, as an alternative explanation for the pain-related behavior, is not applicable, indicating that this induction method is useful for modeling pain in rats.
Ameliorating Huntington’s Disease with CRISPR/Cas9
Huntington’s disease is a neurodegenerative disorder that is caused by a defect in the Huntingtin gene (HTT) due to a CAG/glutamine repeat that encodes a polyglutamine (polyQ) repeat in the N-terminal, ultimately leading to a vast array of cellular dysfunctions.
Currently, there is no cure for this disorder. Major efforts are directed towards identifying a way to suppress mutant HTT (mHTT) expression as a way to inhibit disease manifestation association through the CAG repeat. However, animal studies have shown that this is a complex task, since the huntingtin gene is associated with development and its absence can lead to embryonic lethality.[3]
Using HD140Q-KI mice to model Huntington’s disease, genetic manipulations using CRISPR can be applied. These mice are known to have nuclear accumulation of mHTT and age-dependent motor deficits.[4]
An experiment by Yang et al. utilized these KI mice and CRISPR in order to study how mHTT could be targeted using CRISPR gene-editing. HD140Q-KI mice were either injected with control-gRNA or HTT-gRNA. The control-gRNA mice demonstrated deteriorating motor abilities, as was expected, while the HTT-gRNA mice demonstrated increased abilities across the 3-month study period.
While the two groups performed equally during the start of the experiment, significant differences in motor abilities were observed by the end. By the 6-week mark, the HTT-gRNA mice were able to stay on the Balance Beam for as long as the wild-type mice, indicating that their motor abilities had improved from the baseline. On the Rotarod, the HTT-gRNA mice became comparable to wild-type mice by the 9-week mark.
These findings demonstrate how CRISPR can offer therapeutic solutions and possibilities for diseases that have a genetic component that affects behavior.
Using CRISPR/Cas9 to Understand Astrocytes Function
The CRISPR/Cas9 system can also be used to identify and/or explore the role of known brain structures in order to get a clearer understanding of their function.
Astrocytes, for example, even though they have a clear role in neural information processing, remain elusive in terms of their role in regulating behavior. In terms of structure, astrocytes are known to express circadian clock genes in vitro. Thus, it is logical to hypothesize that astrocytes may be involved in regulating daily rhythms and behaviors. However, there is much left to be determined.[5]
Using CRISPR/Cas9 gene-editing, it is possible to ablate the clock gene Bmal1 specifically localized to astrocytes found in the suprachiasmatic nucleus (SCN), an area of the hypothalamus that is involved in regulating circadian rhythms, and subsequently study the effects on behavior.
When these mice are injected with the necessary materials for CRISPR-induced loss of Bmal1 in astrocytes, the mice’s locomotion levels are significantly affected. As a result, Bmal1-depleted mice show lengthened locomotor activities compared to controls with normal Bmal1 levels as determined by cage activity behaviors such as Wheel Activity.
This example demonstrates the specificity that the CRISPR/Cas9 technique has to offer for behavioral scientists.

Modeling Fragile X Syndrome in Rats with CRISPR
Mutations that inactivate the FMR1 gene are associated with Fragile X Syndrome (FXS), a type of neurodevelopmental disorder. Symptoms associated with FXS include symptoms of mild to moderate intellectual disability or retardation. Usually, FXS patients have problems with their short-term and working memory, language skills, executive function, and visuospatial abilities.
Currently, existing FXS animal models show mixed results when it comes to the efficacy of modeling FXS. For example, the most popular FXS model, the FMR1 knockout (KO) mouse, which is generated by manipulating either exon 1 or exon 5 along with the FMR1 gene’s promoter region, will still show highly variable behavioral phenotypes.
However, this model has many disadvantages when it comes to producing the global cognitive dysfunction observed in the FXS clinical phenotype. The traditional models show almost normal behavior in the Morris Water Maze test and have normal working memory in the Radial Arm Maze test when compared to wild-type mice.
CRISPR/Cas9 technology can be used to generate FMR1 knockout rats through disrupting the FMR1 gene’s fourth exon. The results are histologically and behaviorally significant as this model has impairments in long-term synaptic plasticity, leading to compromised hippocampus-dependent learning and diminished cognitive skills. In the next section, we will see how CRISPR/Cas9 gene-edited FMR1exon4-KO rats behave. [6]
Assessing Behavior in FMR1 Rats
Behaviorally, these rats show significant deficits in their performance during the probe trial of the Morris Water Maze test.
In order to rule out the possible explanation that motor dysfunction contributes to the learning difficulties observed in the Morris Water Maze test, these rats can be subjected to locomotor activity such as the force-plate actometer.
These rats display diminished social interaction when tested in the Three-Chamber Test. In the first part of the test, the experimental rats do well but fail in the second and final part.
When presented in a situation where they can spend time with an object or a stranger/object rat, FMR1exon4-KO rats will pick the stranger/novel rat over the object. This preference is also seen in normal wild-type mice. However, when these rats are again presented with the same ‘stranger’ rat as before (which by now they are familiarized with them) and another new rat, they spend more time with the rat they encountered previously (that was present initially during the trial with the object).
Such a preference is abnormal since that is not how normal wild-type rats behave. Wild-type rats spend significantly more time with the newly presented rat than with the rat they have encountered previously, indicating higher sensitivity to social interaction and social novelty.
All in all, the CRISPR/Cas9 method produces a model that captures the key features of FXS in a manner that is more consistent than what existing models can do.
Using CRISPR in Mice to Induce Rett Syndrome and Bioluminescence Imaging
Rett syndrome (RS) is a neurodevelopmental disorder that has genetic causes. It is rare but serious since it affects brain development, ultimately leading to both physical and mental disability. RS patients, in addition to demonstrating cognitive defects, may also have circadian clock dysfunction. Since RS patients exhibit sleep-related problems and abnormal sleep patterns, it’s probable that the circadian clock is somehow gets affected.
To model RS in mice, mice that are deficient in Methyl-CpG-binding protein 2 (Mecp2) are used, since a mutation in Mecp2 in humans is associated with RS. Mecp2 is a gene that encodes for a methylated DNA-binding nuclear protein. However, this approach is limited since the canonically engineered mice display such severe neurological problems and consequently have a short life span, making it challenging to conduct research.
In order to study the mouse behavior as it pertains to circadian clock dysfunction specifically within an RS disease model, researchers can use the CRISPR gene-editing method to specifically target the RS disease genome in a focused and strategic manner.[7] In the next section, we will take a look at how this gene-editing technology can be used to create an RS model that is designed to give insights on circadian rhythms, too.
Studying Circadian Rhythm in Rett Syndrome Mice
The Periodic Circadian Regulator 2 (PER2) gene is specifically expressed as a part of the circadian pattern in the brain’s suprachiasmatic nucleus. Thus, it is a gene that is of interest in research that deals with circadian rhythms.
In order to study the circadian rhythms within the Rett Syndrome disease model, it is possible to generate an RS mouse model with Mecp2-deficient mice by using wild-type C57BL/6 mice as a background. Compared to wild-type mice, Mecp2 mutants display locomotor deficiency as quantified by Infrared Motion Sensors. Mecp2 mutants have lower activity levels across an entire 24-hr period. This low activity patterns remains even when binning the hours into two 12-hr periods and comparing the bins with each other.
In order to ensure that the observed effects were not due to neurological symptoms, it is possible to use CRISPR/Cas9 editing to knock-in the circadian gene PER2Luciferase(PER2Luc). Thus, the Mecp2 mutant PER2Luc knock-in mice model has the PER2 gene fused together with a luciferase reporter, enabling real-time monitoring of circadian oscillation of PER2 expression through bioluminescence imaging.
CRISPR/Cas9-edited Mecp2 mutant mice do have lower levels of the circadian gene PER2, as established through imaging using the luciferase reporter. This strongly suggests that the behavioral changes occurring (decreased locomotion and activity) are due to issues with the circadian rhythms.
This example demonstrates that complex and informative mouse models can be created through the CRISPR/Cas9 system which are useful and efficient for following a hypothesis along every step of the way.
Conclusion
The amount of behavioral studies done with mice and rats using CRISPR/Cas9 technology is on the rise due to the high specificity that this approach provides.
In many cases, such as with model induction, using CRISPR genome-editing techniques is easier than the alternative. For example, this is especially clear with cancer models which typically require many laborious steps and time-consuming approaches. [8]
Also, as a technique for model induction, CRISPR genome-editing addresses and resolves many of the pitfalls of existing animal models, as mentioned previously for Fragile X and Rett syndromes, for example.
In fact, CRISPR may even be the technique of the future, especially since in July 2019, it was featured in the news for receiving the green light in the USA to be used in human participants to study whether inherited blindness can be cured.
Thus, this technique, in addition to serving behavioral research, may be used as a therapeutic means as well and deserves the attention of researchers in the field of behavioral neuroscience.
References
- Barrangou, Rodolphe. “The roles of CRISPR–Cas systems in adaptive immunity and beyond.” Current opinion in immunology 32 (2015): 36-41.
- Moutal, Aubin, et al. “CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide.” Pain 158.12 (2017): 2301.
- Rubinsztein, David C. “Lessons from animal models of Huntington’s disease.” TRENDS in Genetics 18.4 (2002): 202-209.
- Yang, Su, et al. “CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease.” The Journal of clinical investigation 127.7 (2017): 2719-2724.
- Tso, Chak Foon, et al. “Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior.” Current Biology 27.7 (2017): 1055-1061.
- Tian, Yonglu, et al. “Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats.” Frontiers in molecular neuroscience 10 (2017): 269.
- Tsuchiya, Yoshiki, et al. “Disruption of Me CP 2 attenuates circadian rhythm in CRISPR/Cas9‐based Rett syndrome model mouse.” Genes to Cells 20.12 (2015): 992-1005.
- Mou, Haiwei, et al. “Precision cancer mouse models through genome editing with CRISPR-Cas9.” Genome medicine 7.1 (2015): 53.