
The intricate world of pathogens and diseases is expanding at an alarming rate. All pathogens, no matter how bizarre, follow the same patterns of replication. Previously, pathogens were regarded as organisms that could simply replicate themselves whilst causing harm to a living cell or body. This process of replicating themselves has a fundamental requirement; a genome, whether it be DNA or RNA. This was considered universally true for all pathogens until the discovery of prions in 1982 by Stanley B. Prusiner. For which he was awarded a Nobel Prize in Physiology or Medicine in 1997.
The word, “prion” is derived from “proteinaceous infectious particle”. It defines a mysterious class of infectious agents that cause several neural degenerations in mammals. In humans, these particularly include the Creutzfeldt-Jakob disease (CJD), Kuru and Gerstmann-Straussler-Scheinker Syndrome.
A prion, simply put, is a protein that has attained an abnormal conformation that enables it to cause disease.[1]
In a world where all pathogens required nucleic acids for replication, prions were the infectious agents of science fiction. Since their discovery, prions have baffled scientists with their ability to replicate despite the lack of a nucleic acid. The reason why prions are interesting is that they challenge the central dogma of molecular biology. This has been called the “protein-only hypothesis” which is a theoretical model that explains how a protein replicates on its own without nucleic acid.[2]
Another crucial, yet, interesting fact about prions is their transmissibility. Prion that causes disease in one species cannot do the same in another. This is called a species transmission barrier. Research and particularly, the development of mouse models becomes difficult due to this limitation. Despite this, cross-species infections are still seen in diseases such as Bovine spongiform encephalopathy (BSE) and CJD.
Pathophysiology of Prion Disease
Before going into the details of various mouse models developed for prion disease research, it is important to understand the pathology of prion disease.
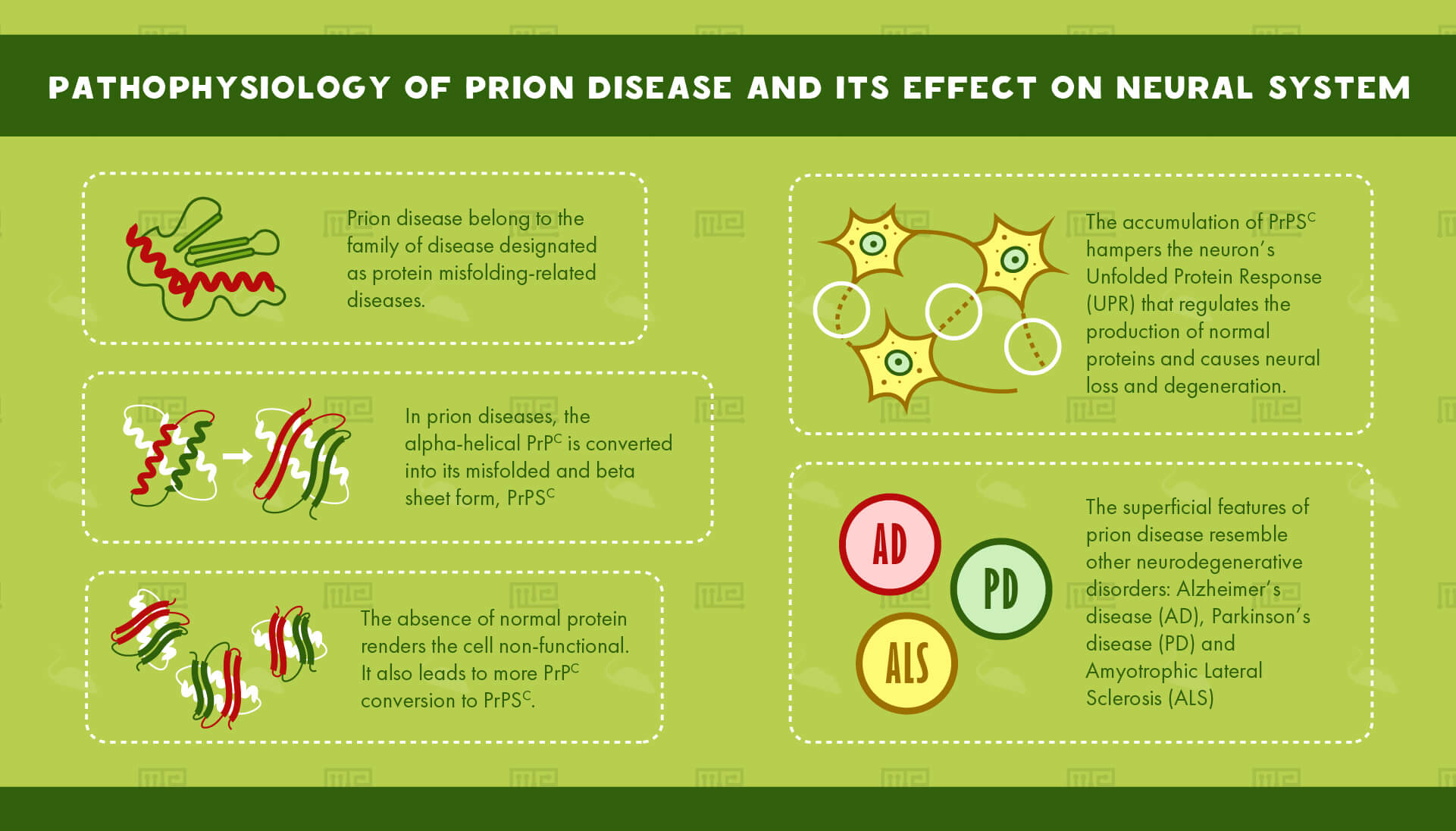
Prion disease belongs to the family of diseases designated as protein misfolding-related diseases. These involve aggregation of abnormal proteins in cells and tissues.
The culprit behind these mysterious diseases is a protein commonly found in cell membranes: The Prion ProteinC or PrPC, an alpha-helical protein of 35-36 kDa size. It is coded by the gene, PRNP. PrPC is found in all tissues of the body but it is most abundantly found in the Central Nervous System (CNS). It is primarily involved in cell adhesion as well as cell-cell interactions in the brain. It is also necessary for normal synaptic function.[3]
In prion diseases, the alpha-helical PrPc is converted into its misfolded and beta sheet form, PrPSC. The accumulation of PrPSC hampers the neuron’s Unfolded Protein Response (UPR) that regulates the production of normal proteins. The absence of normal protein renders the cell non-functional. It also leads to more PrPC conversion to PrPSC. This accumulation causes neural loss and degeneration.
The superficial features of prion disease resemble other neurodegenerative disorders. Alzheimer’s disease (AD), Parkinson’s disease (PD) and Amyotrophic Lateral Sclerosis (ALS), all involve aggregation of misfolded proteins.[4]
Exploring and studying prion disease
Prions’ lack of nucleic acid poses a challenge for scientists to replicate them in the laboratory. This complicates prion research and hinders the development of therapy against prions. Despite this, many in-vitro assays, ex vivo models, and animal models have been developed to help scientists understand the dynamics of prion disease.
Cell-based In vitro Assays
Cell lines and cell-based in vitro models are the most widely used tools for studying the cell biology of prion amplification. Cell-based models are created by infecting a specific type of cells with prion strains. For example, the cell line, N2a is a commonly used prion cell line. N2a is mouse neuroblastoma cells infected with mouse prion strains. Similarly, GT-1 is another cell line created to produce mouse prion strains.[5]
There are at least 15 neural and non-neural cell lines for prion research in existence.[5] They have multiple advantages over ex vivo and animal models. They are low cost, simple and can be easily reproduced and standardized. However, these cell lines also have a disadvantage. The effects of prion disease on cell-based models don’t always correspond to the human disease.
In November 2017, a team of scientists from the University of Edinburgh successfully replicated three prion strains in astrocytes developed from human pluripotent stem cells. This is the first cell-based prion replication system developed from human stem cells.[6]
Ex vivo Models
Ex vivo models involve the extraction of tissues from live organisms which are then transferred to an artificial environment. This is an effective technique to assess prion replication. One such ex vivo model is the organotypic slice culture. In particular, the murine cerebellar organotypic slice culture is a popular choice.[5]
Organotypic slice culture involves extraction of thin slices from different parts of the brain. These slices are typically used to study the stages of neural development. Murine cerebellar organotypic slice culture is the in vitro culturing of slices obtained from mouse cerebellum. These slices are then infected with prion strains to observe the changes that occur due to prion infection.[7]
This model is used for studying PrPC to PrPSC conversion, interactions, neural toxicity, and also for studying the effects of new therapeutic approaches. With regards to their scope and complexity, organotypic slice cultures are intermediate to in vitro assays and in vivo animal models. However, their advantage over in vivo animal models has yet to be identified as animal models provide a better picture of prion disease neuropathology and biochemistry.
Synthetic Recombinant PrpC
The isolation and purification of prion particles is a challenging process. Originally, Stanley Prusiner purified prion particles by fractionating hamster brain homogenates and then checking them for infectivity. This was a long and tiresome process. Presently, scientists have managed to synthesize recombinant PrP (recPrP) in Escherichia coli (E.coli) expression systems. It is an effective tool for studying the physiological function of PrP as well as its pathogenicity and structural properties. Human recombinant PrP with various structural modifications has been produced to study the pathogenicity of different PrPC structures.
Although recPrP has made prion particle production considerably easy, obtaining quality titers is still a challenge. Studies have shown that recPrP does not yield significant infectivity rates in inoculated mice.[8] Moreover, the prion particles are not suitable for structural characterization.[9]
Low infectivity of bacterially produced recPrP can be linked to a number of reasons. Efficient prion production may require PrP that is eukaryotically expressed and post-translationally modified. It may also require certain cofactors.[9] Studies show that efficient in vitro production of high infectivity prions requires RNA and phosphatidylethanolamine (PE).[10]
Computational Biology and Prion Disease Research
In silico modeling has aided the development of simulation methods for studying the manner of interaction between various molecules, including the determination of binding affinity and binding modes. Bioinformatics approach has also made it possible to identify specific genes that affect prion disease pathogenicity and incubation times.
Specific protein-protein interaction networks have also been constructed using computational methods. These networks aid in observing the effects of specific genes and anti-prion compounds on protein interactions that influence prion disease pathogenicity.[11]
Computer modeling has enabled scientists to determine the interactions and effects of novel therapeutics and anti-prion compounds.
This approach towards prion disease research has also helped in identifying various diagnostic markers that can prevent prion infections by early detection of prion particles in infected blood or meat.
In spite of the valuable contributions of computational biology, in silico models are not completely reliable and the results are required to be substantiated by wet lab experiments.
Mouse Models: A breakthrough technique in Prion Research
Experiments to understand prion transmission was initially conducted on wild-type and inbred mice. But the development of transgenic mouse models has revolutionized prion research. The first ever transgenic mouse model was created in 1989 to understand prion strains and transmission barriers. It was the same transgenic mouse model that helped observe the effects of amino acid sequence changes in the Prnp gene on the model’s susceptibility to prion strains.
Conventional models
Wild-type inbred mice
Before scientists started using transgenic mouse models for studying prion disease, wild-type inbred mice were the most popular choice. C57Bl/6L, RⅢ, and CD1 are amongst the most widely used inbred mice models.
Inbred mouse models were used to produce and transmit prion strains, particularly those of Scrapie. For example, the inbred mouse strain Me7 was created by the direct transmission of Scrapie of Suffolk sheep into mice.
Inbred strains of wild-type mice explained the transmission and replication of human prions.
This model is ideal for testing anti-prion compounds against Scrapie, a prion disease in sheep. As an advantage, they have a consistent and controlled genomic background. Yet, inbred mice cannot be infected with human prions. Hence, they cannot be used to reproduce the effects of human prion disease.[5]
PrP knockout mice; Prnp0/0
Prnp knockout mice do not express the Prion protein (PrP) because major portions of the Prnp gene have been deleted. Studies involving the PrP knockout mouse strain, Ngsk Prnp0/0 have shown that deleting portions of the Prnp gene result in neural degeneration. The deletion of a splice acceptor located on intron 2 of the Prnp gene caused overexpression of a gene called Doppel or Dpl. In the Ngsk Prnp0/0 mouse’s brain, Dpl is upregulated due to the high activity of the Prnp promoter. Various studies have linked the overproduction of Doppel to neural degeneration in PrP knockout mice.
This model does not reveal any similarities with human degenerative diseases. But, it does explain the phenotype produced due to lack of the PrP protein.
Transgenic Mouse Models of Prion Disease
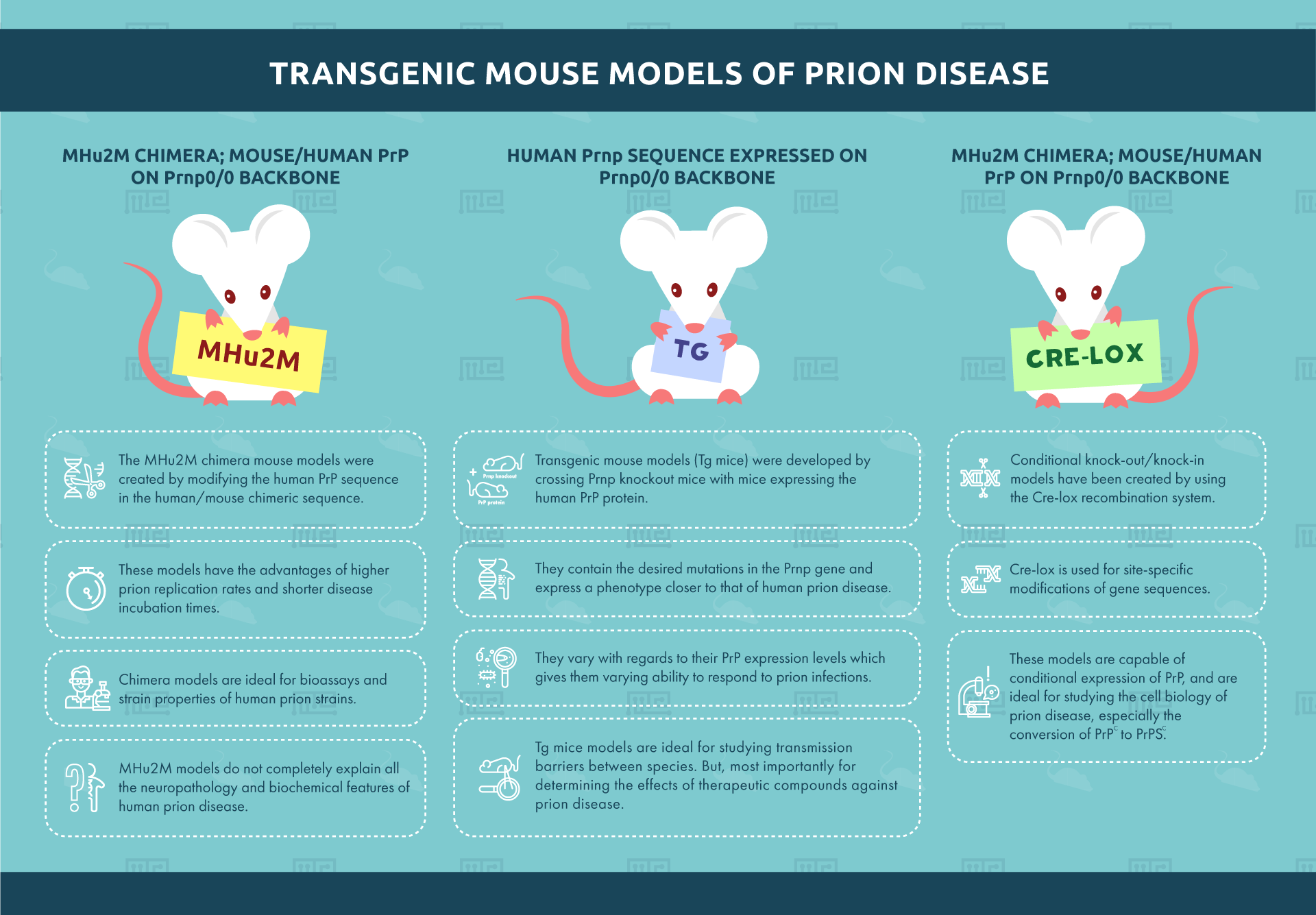
Human Prnp sequence expressed on Prnp0/0 backbone
Transgenic mouse models (Tg mice) were developed by crossing Prnp knockout mice with mice expressing the human PrP protein. These transgenic mice contain the desired mutations in the PRNP gene and express a phenotype closer to that of human prion disease. Around 45 transgenic mouse lines currently exist.[5] They vary with regards to their PrP expression levels which gives them varying ability to respond to prion infections.
Tg mice models are ideal for studying transmission barriers between species. But, most importantly for determining the effects of therapeutic compounds against prion disease. However, the varying expression levels of PrP complicate the selection of a suitable model.
MHu2M Chimera; mouse/human PrP on Prnp0/0 backbone
The MHu2M chimera mouse models were created by modifying the human PrP sequence in the human/mouse chimeric sequence. The sequence construct contains less human and more mouse PrP sequences. These models have the advantages of higher prion replication rates and shorter disease incubation times. Chimera models are ideal for bioassays and strain properties of human prion strains. Unlike the Human Prnp/Prnp0/0 model mentioned above, MHu2M models do not completely explain all the neuropathology and biochemical features of human prion disease.
Conditional knockout/knockin models
Conditional knock-out/knock-in models have been created by using the Cre-lox recombination system. Cre-lox is used for site-specific modifications of gene sequences. These models are capable of conditional expression of PrP. These are ideal for studying the cell biology of prion disease especially the conversion of PrPC to PrPSC.
Other Mouse Models of Prion Disease
Models with full-length PrP sequence on Prnp0/0 backbone
Mice with full-length PrP sequence introduced in the Prnp0/0 backbone are efficient models for studying the prion spread, incubation times and neurotoxicity. Prnp null mice are immune to prion infections due to the lack of PrP. But the introduction of a full-length Prnp sequence reinstates prion disease in Prnp null mice.
The full-length sequence models are ideal for studying the prion pathogenicity, neurotoxicity, incubation time and transmission rates of prion diseases.
However, the reintroduction of mouse Prnp gene makes this model less suitable for human prion disease research. This is so because this model does not completely explain the biochemistry and neuropathology of human prion disease.
Models with deletions and point mutations in PRNP gene
Mouse models with deletions and point mutation in the PRNP gene were initially created to understand human Inherited Prion Disease (IPD).
Studies have shown that in-frame deletions and mutations lead to progressive cerebellar degeneration in mice. This may result from interactions between Doppel protein and truncated PrP or because of formation of PrP heterodimers.[12]
These models are ideal for studying the pathophysiology of prion diseases. However, like the previously mentioned one, this model also does not completely explain the biochemical aspects of human prion disease.[5]
Behavioral Testing in Mouse models of Prion Disease
Behavioral studies in Transgenic mouse models
Knockout mouse models are widely used to study the function of the PrPC protein and how it causes prion diseases. In order to completely understand prion diseases, various behavioral assays have been developed.
Mouse Strains in behavioral testing
Prion disease behavioral tests are carried out using several strains of mice. These are:
- C57BL, C57BL/6
- A2G
- VL Mice
- VM Mice
- C57BL/10J
- Nya:NYLAR Mice
- Mice inoculated with Scrapie strains, Me7, 22l, 22A and 79-A
- CD1 Mice
- Zurich 1 Mice (Transgenic)
Out of all the above-mentioned strains, C57BL/6, C57BL/10J, CD1 and Zurich 1 mouse strains are most commonly used.
Morris Water Maze (MWM) Test
The water maze test, also known as the Morris Water Maze (MWM) is used to measure the subject’s spatial memory and the desire to explore their surroundings.
The first knockout mouse, Zurich 1 was subjected to the water maze test to determine any behavioral changes during a prion infection. The mice showed no change in spatial recognition.
Y Maze and Two-Way Active Avoidance Test
The Y maze is used to determine the animal’s spatial recognition and memory. It also gives insight into the animal’s exploration ability.
The same set of Zurich 1 knockout mice from the MWM was tested through the Y maze discrimination and two-way active avoidance tests. Like the Morris Water Maze test, the mice did not show any abnormality in memory, spatial learning and problem-solving.[13]
Open Field Test
Open Field Locomotion Test is conducted to monitor any changes in habituation learning over time. The open field test has shown that knockout mice models suffer from memory and problem-solving difficulties.[14]
Buried Food Test
The buried food test is conducted to observe changes in the subject’s olfactory senses. It determines the subject’s ability to remember and recognize various smells. Studies show that Zurich 1 mice suffer from sensory abnormalities.[15]
Elevated Plus Maze
Elevated plus maze helps measure the subject’s exploratory ability as well as the level of anxiety. The maze has two distinct zones: closed arms and open arms. The lighting in the open arms is changed regularly to observe whether the subject chooses to explore or not.
Open field and elevated plus maze tests have shown that the PrPc overexpression mouse model performs better with regards to anxiety and exploration. [14]
Conclusion
Neurodegenerative diseases do not always have apparent symptoms. Prion diseases, in particular, cannot be diagnosed earlier because of their long incubation time. In this context, mouse models and behavioral testing can help scientists figure out behavioral cues to diagnose prion diseases. This will ultimately give scientists a better understanding of the dynamics of prion disease and help discover novel therapeutics.
References
- What Is a Prion? (n.d.). Retrieved from https://www.scientificamerican.com/article/what-is-a-prion-specifica/
- Ma, J., & Wang, F. (2014, 08). Prion disease and the ‘protein-only hypothesis’. Essays In Biochemistry, 56, 181-191. doi:10.1042/bse0560181
- Collinge, J., Whittington, M. A., Sidle, K. C., Smith, C. J., Palmer, M. S., Clarke, A. R., & Jefferys, J. G. (1994, 07). Prion protein is necessary for normal synaptic function. Nature, 370(6487), 295-297. doi:10.1038/370295a0
- Fernández-Borges, N., Eraña, H., Venegas, V., Elezgarai, S. R., Harrathi, C., & Castilla, J. (2015, 09). Animal models for prion-like diseases. Virus Research, 207, 5-24. doi:10.1016/j.virusres.2015.04.014
- Brandner, S., & Jaunmuktane, Z. (2017, 01). Prion disease: Experimental models and reality. Acta Neuropathologica, 133(2), 197-222. doi:10.1007/s00401-017-1670-5
- NC3Rs. (n.d.). Retrieved from https://www.nc3rs.org.uk/news/cell-based-model-means-step-forward-creutzfeldt-jakob-disease-research
- Falsig, J., Sonati, T., Herrmann, U. S., Saban, D., Li, B., Arroyo, K., . . . Aguzzi, A. (2012, 11). Prion Pathogenesis Is Faithfully Reproduced in Cerebellar Organotypic Slice Cultures. PLoS Pathogens, 8(11). doi:10.1371/journal.ppat.1002985
- Collinge, J., & Clarke, A. R. (2007). A general model of prion strains and their pathogenicity. Science, 318(5852), 930-936.
- Schmidt, C., Fizet, J., Properzi, F., Batchelor, M., Sandberg, M. K., Edgeworth, J. A., … & Linehan, J. M. (2015). A systematic investigation of production of synthetic prions from recombinant prion protein. Open biology, 5(12), 150165.
- Supattapone, S. (2014). Synthesis of high titer infectious prions with cofactor molecules. Journal of Biological Chemistry, 289(29), 19850-19854.
- Hwang, D., Lee, I. Y., Yoo, H., Gehlenborg, N., Cho, J. H., Petritis, B., … & Price, N. D. (2009). A systems approach to prion disease. Molecular systems biology, 5(1), 252.
- Fischer, M., Rülicke, T., Raeber, A., Sailer, A., Moser, M., Oesch, B., … & Weissmann, C. (1996). Prion protein (PrP) with amino‐proximal deletions restoring susceptibility of PrP knockout mice to scrapie. The EMBO journal, 15(6), 1255-1264.
- Lipp, H., Stagliar-Bozicevic, M., Fischer, M., & Wolfer, D. P. (1998, 09). A 2-year longitudinal study of swimming navigation in mice devoid of the prion protein: No evidence for neurological anomalies or spatial learning impairments. Behavioural Brain Research, 95(1), 47-54. doi:10.1016/s0166-4328(97)00209-x
- Lobaosoares, B., Walz, R., Carlottijr, C., Sakamoto, A., Calvo, F., Terzian, A., . . . Bianchin, M. (2007, 10). Cellular prion protein regulates the motor behaviour performance and anxiety-induced responses in genetically modified mice. Behavioural Brain Research, 183(1), 87-94. doi:10.1016/j.bbr.2007.05.027
- Pichon, C. E., Valley, M. T., Polymenidou, M., Chesler, A. T., Sagdullaev, B. T., Aguzzi, A., & Firestein, S. (2008, 12). Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nature Neuroscience, 12(1), 60-69. doi:10.1038/nn.2238